Cyclo[18]carbon (C18) as Electron Acceptor for Organic Solar Cells Applications
Main Article Content
Article Sidebar

 https://orcid.org/0000-0001-9619-0176
https://orcid.org/0000-0001-9619-0176
Haseena Prakash Maiyelvaganan
Abstract
Organic solar cells (OSCs) can be a theoretically promising technology for providing clean and renewable energy. The significant advantages of OSCs compared to their counterparts are low-cost, lightweight, flexibility.[1] However, efficiency limitations, as well as long-term reliability, are major concerns. The design and development of new non-fullerene acceptors (NFAs) (used as electron acceptors in organic solar cells) is currently a major focus of the research. Several NFAs, such as fused ring aromatic cores with strong electron acceptors and rylene diimide-based materials, have shown promising power conversion efficiencies (PCEs).[2] Recently developed molecules ITIC and Y6-based acceptors have shown PCE of more than 18%.[3,4] However, key information related to structure-property relationships, donor-acceptor energy levels matching, and understanding the energy losses are important to improve the efficiencies of devices. In this context, computational studies provide more insights into the molecular level understanding on energy level matching and molecular packing between donor and acceptor materials.
In this investigation, we computationally study whether the recently synthesized Cyclo[18]carbon (C18) molecule can be used as the electron acceptor in organic solar cells. Strong π-electron delocalization and high electron affinity make C18 as the electron acceptor.[5] Thus, density functional theory-based methods are used to characterize the electronic properties, aromaticity, and the nature of interactions between C18 molecules. The important criteria which control charge mobility are reorganization energies and electronic coupling between molecules. We have calculated the reorganization energies using state-of-the-art methods. The nature of interaction between C18 molecules is analyzed using symmetry-adapted perturbation theory (SAPT). The results obtained from this analysis are useful to understand the packing between molecules. The calculated reorganization energies and electronic couplings have shown good charge transport properties.
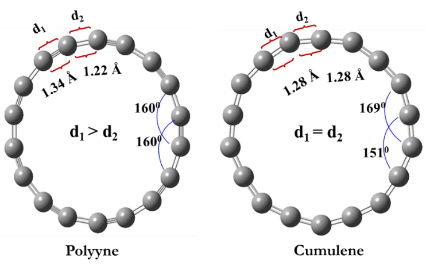
Fig.1. Optimized geometry of polyyne and cumulene structure along with bond angle and distance using CAM-B3LYP/6-311++G (2d,p) method.
The optimized geometries of monomers are shown in fig. 1, along with the bond length data obtained from three different DFT methods. For this study, selecting a suitable DFT method is very important to identify the correct bond length and electronic structure of these molecules. In this study, we have demonstrated the importance of long-range corrected DFT methods for the proper description of highly delocalized bonds.
The aromatic behavior of molecules depends on the nature of bonding (σ and π bonds) between carbon and carbon (C-C) atoms. From our DFT simulations we found the double aromatic nature of C18 rings. The delocalization of two scaffolds of π-electrons oriented towards in-plane (πin) and out-of-plane (πout). Thus, there is a double aromaticity behavior in the circular form of C18 molecules. The bond order differences in polyyne and cumulene rings reflect through aromatic character.
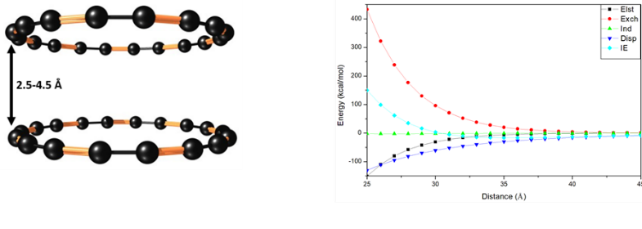
Fig.2. Graphical representation of energy decomposition analysis of two C18 rings at a. eclipsed position change in vertical distance from 2.5Å to 4.5Å. All calculations are carried out using SAPT/aug-cc-pvdz level of theory.
Based on the results obtained from both analyses, we consider only cumulene ring structure for further analysis. The energy decomposition analysis on dimers of C18 is carried out to gain more insights into the nature of interactions between the ring structures and also packing of these molecules in the thin-films. One can see from fig. 2 that the major contribution to the stabilization complex came from the dispersion energy. The large repulsive exchange energy compensates attractive terms. However, it is interesting to note that the large exchange-repulsion indicates the considerable overlap of electron densities. Such a large overlap of electron density might lead to large electronic coupling between rings. We have calculated the electronic coupling values between the rings, and we found (LUMO-LUMO) electronic couplings values in the rages of 400 meV. As discussed before, large electronic couplings and low internal reorganization energies are the key parameters for better charge mobilities. Results obtained from our calculations indicate that the C18 may exhibit better charge mobilities as it shows large electronic coupling with low reorganization energies.
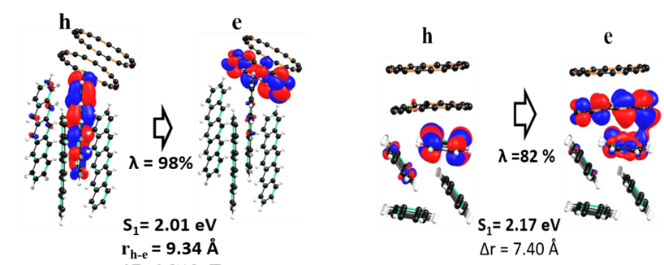
Fig.3. pictorial representation of hole and electron wavefunctions in pentacene-C18 clusters in the lowest excited states.
Further, we have also studied donor-acceptor complexes based on pentacene-C18 clusters as model systems. Time-dependent DFT analysis is carried out to characterize the excited state properties of these clusters. Fig. 3 shows the hole and electron wavefunctions of pentance-C18 clusters in the lowest excited state. The excited state analysis on these clusters reveals that the C18 molecule can be used as acceptor molecules as the lowest excited state has charge transfer in nature (hole localized on pentacene and electron localized on C18). We have studied how molecular packing between donor and acceptor molecules impact the hole and electron delocalization in the excited states. These results give insights into the charge dissociation property of the system of interest. All these results will be useful to provide designing rules for the experimentalist.
How to Cite
Article Details
2. C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder, and X. Zhan, Nat Rev Mater 3, 18003 (2018).
3. C. Cui, Front. Chem. 6, 404 (2018).
4. Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang, and L. Ding, Science Bulletin 65, 272 (2020).
5. A. J. Stasyuk, O. A. Stasyuk, M. Solà, and A. A. Voityuk, Chem. Commun. 56, 352 (2020).